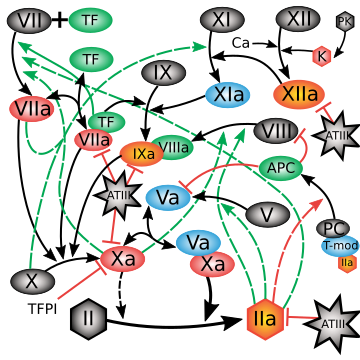
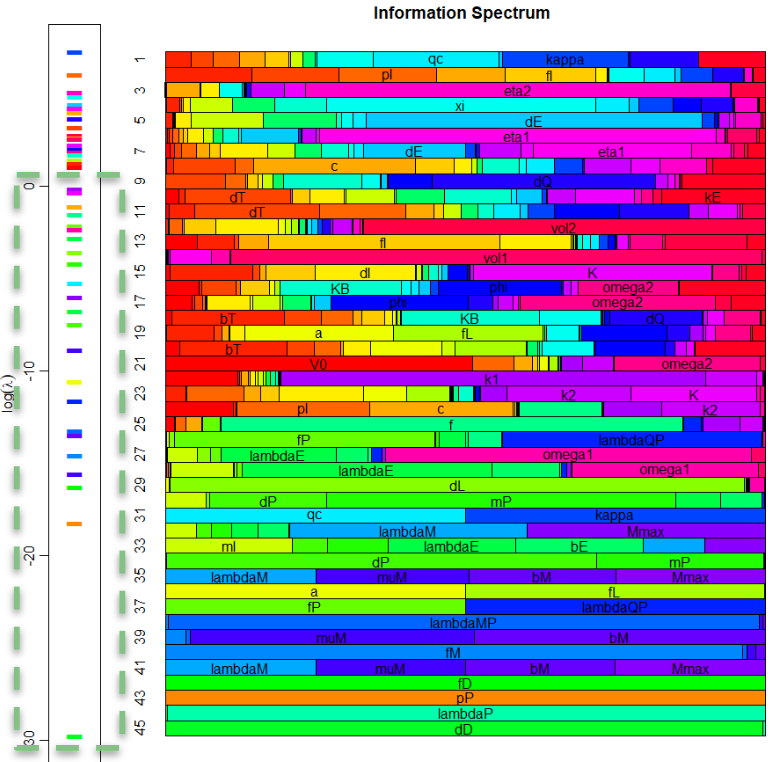
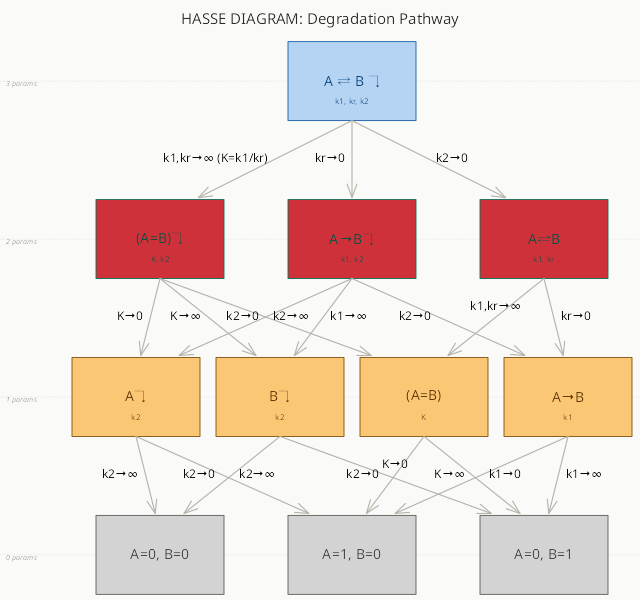
Cross Stream
AI
Home
About
Examples
Contact
Latest Articles
The Coagulation Cascade: Information Geometry in your blood, stream-lined
Understanding AIDS treatment
Information Geometry 101